
Boundary condition
 
Posts: 8617 Joined: 30-Aug-2008 Last visit: 30-May-2026 Location: square root of minus one
|
Thanks for sharing your results, Infundibulum. I had read that high pH is very bad for the fungal indoles as well, so perhaps the dark precipitates also included a share of decomposition products. Do you recall whether they might have had a bluish shade? On the basis of your information my thinking on the calcium psilocybate ("Ca-Psopate" ) experiment goes as follows:- Firstly, perhaps Ca-Psopate has an appreciable water solubility after all? Secondly, in order to check the assertion that it is possible to precipitate Ca-Psopate, a source of calcium with much lower pH must be used. I would suggest calcium acetate as a suitable alternative. Using a highly soluble calcium salt should increase the chance of precipitating Ca-Psopate through the common-ion effect. Thirdly, I would consider investigating the effect of filtering the mushroom tea through a layer of calcium carbonate. This, of course, is beginning to resemble chromatography  I suppose I ought to get experimenting myself. Now, where did all that psilocybin go...? “There is a way of manipulating matter and energy so as to produce what modern scientists call 'a field of force'. The field acts on the observer and puts him in a privileged position vis-à-vis the universe. From this position he has access to the realities which are ordinarily hidden from us by time and space, matter and energy. This is what we call the Great Work." ― Jacques Bergier, quoting Fulcanelli
|
|
|
|
|

❤️🔥

Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Would one possible way to interpret results from Infundibulum be that some Ca-Psilocibinate did precipitate but was not bioavailable? That could explain why the total activity of the combined precipitate + solution was low (which is how I understand the info at this time). Or are we sure that Ca-Psilocibinate is bioavailable? Someone may know. This article studied a different low solubile Ca salt (CaCO3) and saw that adding vitamin D helped it's bioavailability. Commercial CaCO3 supplements are sold with vitamin D. Taking them with a full stomach may also help, based on the recommendations coming from the Ca supplement industry. We may want to consider adding vitamin D to some of our future bioassays based on Infundibulum's previous work if that was not done already. I've also come across darkish precipitate. Seemed to happen at pH~11. Precipitates at lower/higher pH did not seem as dark. I've attached a picture of the dark precipitate. So far I have not done a bioassay of the parts that are separating out an/or a final neutral solution. downwardsfromzero, I'm planning on trying CaCl2 to check the common ion effect. Picked up a small 1lb bag at the local brewery shop. I'm not aware of any specific high pH concern for psilocybin, anyone have more info on that? First attached paper has folks working at pH 11.5, no peak degradation for pure psylocybin after a short time (but longer time or mushroom components could change that). Light can be a big deal (second attachment) so minizing light exposure when working with mushroom extracts is a good practice I think.
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Ok, after adding 20% CaCl2 to the solution that had been brought to a high pH woth Ca(OH)2 more stuff started to "coagulate" in the solution. The solution lost its cloudiness too. The coagulated stuff was separated and turned dark during drying. Some stuck to the jar walls and water was added to see if it was soluble in fresh waster (it was not). This was to see if the coagulate was only Ca(OH2) So at this point we have stuff precipitating out as the pH is raised with Ca(OH)2 and also stuff that clumps together when CaCl2. Based on previous work by Infundibulum, the first set of precipitates may not be very active. My pH meter seems to be on the fritz, but as best I could tell, the pH went down after CaCl2 was added. Will keep on working on this. At some point will begin bioassays. Input welcome. Pictures post CaCl2 addition below (coming out of solution, then in fresh water, and finally in the filter). Loveall attached the following image(s):  IMG_20180318_104730967.jpg (3,275kb) downloaded 481 time(s). IMG_20180318_113119884.jpg (2,736kb) downloaded 478 time(s). IMG_20180318_161116047.jpg (3,773kb) downloaded 478 time(s).
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Ok, another way to spearate precipitates can be done by basifying with Ca(OH)2, adding ammonium sulfate, and adding acetone. The acetone separates in this salty condition and a discting layer of white stuff forms near the bottom of the acetone layer (image attached). What does this mean? Nothing new I think,essentially we got tons of mushroom junk as established before and there are different ways to get stuff to precipitate. Current strategy I'm trying is to precipitate stuff in different ways and figure out where most the actives end up. Seems like they prefer the water so far, if so, after many cleanups one can try to dry the water to a solid residue and either (1) pickup the actives with denatured alcohol (which is what I have acess to) or some kind of dry solvent, or (2) directly make a concentrated tincture or pills with the dry residue. Loveall attached the following image(s): IMG_20180319_082327164.jpg (3,833kb) downloaded 458 time(s).
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Apparently ammonium sulfate can be crashed out of solution by adding alcohol (methanol/ehtanol/IPA). Anyone have experience with this? If so, one possible simple path is, 1) Dilute ammonia mushroom extract, filter, reduce, filter. 2) Add accetone, let settle and filter. 3) Add ammonium sulfate and allow layers to separate. Recover water layer and filter if needed. 4) Add alcohol: Ammonium sulfate (and other stuff?) crashes out, filter that out. 5) Resulting ammonia, alcohol, water should be pretty clean and *may* contain most of the actives. Dry to obtain ammonium psilocobinate and any surviving mushrooms stuff. Any thoughts or obvious issues with this before trying ot out? Thanks.
|
|
|
Boundary condition
Posts: 8617 Joined: 30-Aug-2008 Last visit: 30-May-2026 Location: square root of minus one
|
Just chiming in with words of support here! Unless you've got an unlimited supply mushrooms, perhaps you might want to try a dry run of the process (that is, still with water but missing out the mushrooms, of course) just to see what happens when each of the solvents etc. gets added. And/or try it on the test tube scale if you haven't been already. ^^ Quote:pickup the actives with denatured alcohol Food grade surely would be better. It would be a shame to ruin the extract at the last hurdle, so to speak. “There is a way of manipulating matter and energy so as to produce what modern scientists call 'a field of force'. The field acts on the observer and puts him in a privileged position vis-à-vis the universe. From this position he has access to the realities which are ordinarily hidden from us by time and space, matter and energy. This is what we call the Great Work." ― Jacques Bergier, quoting Fulcanelli
|
|
|

DMT-Nexus member
Posts: 1104 Joined: 11-Feb-2017 Last visit: 18-Jan-2021
|
Also watching your work closely, Loveall. Cannot help at this point as acid/base chemistry intricacies are beyond my knowledge. I just started my mushroom growth for this season so it will take quite some time before being supplied with enough shrooms to reproduce your steps. But I will do when being able to. Please keep up the great work 
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Thanks for the encouragement guys, it helps. Want to give an update on the ongoing work so far. Looks like it is all about managing the mushroom gunk. Psilocybin may even have an affinity to stick to proteins as mentioned before in other threads. I tested a water extract with 2% ammonium hydroxide (pH ~ 11) working in dim light. Mushrooms turned jet black. After filtering and reducing the pH was slightly acidic (so no need to go for ammonium psilocybinate). There is A LOT of junk in this alkaline water extract, as we found when doing the calcium hydroxide work. Fortunately, ammonium sulfate and/or acetone do a good job at crashing out stuff. After removing what crashes out and drying, a brown ammonium sulphate salt is left. I guess it could be cleaned with acetone (similar to what is done in the Hoffman method but I skipped this step). Collecting this browm salt and doing an denatured alcohol extract (I don't have easy access to methanol) gave a gray cloudy liquid. After drying this liquid a nice scrapable shiny brown dry residue was left. I got about 200mg of it from about 100g of fresh Mushrooms (pictured below). I have not tested the residue yet. Work comtomies and other tests have been done, but what is outlined above is giving the best results to date as far as clean looking scrapable product (but until bioassy is done it may all be for nothing).Based on what I've seen while doing this work, this is what I'm trying next: 1) Very dilute ammonium hydroxide (pH ~ 9) mushroom extract. Additional base needs to be added after adding mushrooms to keep the water pH near 9. Logic here is that too high of a pH seems to give more plant mateial in water, and too low pH may convet psilocybin into psilocin due to enzymatic action. Also, depronating the psylocybin shoul make it more water soluble. This is all speculation. 2) Filter, reduce. pH will be slightly acidic after reducing (this is what happens to me). Hoping the boiling water during reduction has denaturlazided cubensis enzymes that convert psilocybin to psilocin. 3) Add 35% ammonium sulphate and chill. Using food or analytical grade since fertilizer grade may have heavy metals. This salt addition lowers pH to about 5.5. Let sit ovenight or longer in fridge. Note: if wanting to try to smoalk later, targeting a pH of 4 here with vinager may be helpful. Also, pH may influence any protein/psilocybin interactions. 4) Separate out precipitate that forms (this should be protein junk). Again, speculating. 5) Dry and collect brown salt 6) Optional? Wash dry brown salt with dry acetone 7) Extract salt with methanol (if not available use denatured alcohol). Filter out the ammonium sulphate which is insoluble in methanol/ethanol. 8 ) Evaporate the alcohol (remimder that methanol is toxic) This feels like the Hofmann method, except that instead of a Chloroform clean we use an ammonium sulphate precipitation (ammonium sulphate is easier to obtain for mea). Also, at the end we don't extract from the mushroom material, instead we extract from a dry salt residue (and use denatured alcohol if that is easier to obtain vs methanol). Will report back later with any promising scrape results and bioassays. Feedback welcome as always. Loveall attached the following image(s): IMG_20180325_191837763.jpg (3,442kb) downloaded 401 time(s).
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Loveall wrote:Thanks for the encouragement guys, it helps.
Want to give an update on the ongoing work so far. Looks like it is all about managing the mushroom gunk. Psilocybin may even have an affinity to stick to proteins as mentioned before in other threads.
I tested a water extract with 2% ammonium hydroxide (pH ~ 11) working in dim light. Mushrooms turned jet black. After filtering and reducing the pH was slightly acidic (so no need to go for ammonium psilocybinate).
There is A LOT of junk in this alkaline water extract, as we found when doing the calcium hydroxide work. Fortunately, ammonium sulfate and/or acetone do a good job at crashing out stuff. After removing what crashes out and drying, a brown ammonium sulphate salt is left. I guess it could be cleaned with acetone (similar to what is done in the Hoffman method but I skipped this step). Collecting this browm salt and doing an denatured alcohol extract (I don't have easy access to methanol) gave a gray cloudy liquid. After drying this liquid a nice scrapable shiny brown dry residue was left. I got about 200mg of it from about 100g of fresh Mushrooms (pictured below). I have not tested the residue yet.
Work comtomies and other tests have been done, but what is outlined above is giving the best results to date as far as clean looking scrapable product (but until bioassy is done it may all be for nothing).Based on what I've seen while doing this work, this is what I'm trying next:
1) Very dilute ammonium hydroxide (pH ~ 9) mushroom extract. Additional base needs to be added after adding mushrooms to keep the water pH near 9. Logic here is that too high of a pH seems to give more plant mateial in water, and too low pH may convet psilocybin into psilocin due to enzymatic action. Also, depronating the psylocybin shoul make it more water soluble. This is all speculation.
2) Filter, reduce. pH will be slightly acidic after reducing (this is what happens to me). Hoping the boiling water during reduction has denaturlazided cubensis enzymes that convert psilocybin to psilocin.
3) Add 35% ammonium sulphate and chill. Using food or analytical grade since fertilizer grade may have heavy metals. This salt addition lowers pH to about 5.5. Let sit ovenight or longer in fridge. Note: if wanting to try to smoalk later, targeting a pH of 4 here with vinager may be helpful. Also, pH may influence any protein/psilocybin interactions.
4) Separate out precipitate that forms (this should be protein junk). Again, speculating.
5) Dry and collect brown salt
6) Optional? Wash dry brown salt with dry acetone
7) Extract salt with methanol (if not available use denatured alcohol). Filter out the ammonium sulphate which is insoluble in methanol/ethanol.
8 ) Evaporate the alcohol (remimder that methanol is toxic)
This feels like the Hofmann method, except that instead of a Chloroform clean we use an ammonium sulphate precipitation (ammonium sulphate is easier to obtain for mea). Also, at the end we don't extract from the mushroom material, instead we extract from a dry salt residue (and use denatured alcohol if that is easier to obtain vs methanol).
Will report back later with any promising scrape results and bioassays. Feedback welcome as always.
Seems like during step 1) the pH dropped all they way down to 8.3. A small film of blue developed at the top of the solution. During filtering there was a small spill that quickly turned blue (first picture). Higher pH extracts (eg CaOH) don't seem to do this so maybe I let the pH get too low? Adding vitamin C stopped/reversed the blueing (second picture where a dash of Vitamin C was added to the right side). I also had not boiled the solution yet so the enzimes that convert psilocybin to psylocin were intact (gentle pH, no heat). Turns out Ca(OH)2 is not soluble in alchohol as far as I can see, so it should not be extracted at the end when using the denatrued alcohol. So nex iteration would be: 1) Ca(OH)2 hot water extract of ground mushrooms, filter, reduce. 2) 35% cold Ammonium sulphate protein precipitation. 3) Dry and collect salts/solids. 4) Optionally wash salts with dry acetone. 5) Extract dry salts with denatured alcohol (or methanol). 6) Dry alcohol and scrape up final product. Willl move on to more tests. Don't plan to keep on updating all the time (I don't want to saturate this thread with ramblings), will update sporadically with lessons learned. Loveall attached the following image(s): IMG_20180327_082719561.jpg (4,494kb) downloaded 387 time(s). IMG_20180327_082911741.jpg (4,138kb) downloaded 386 time(s).
|
|
|
Boundary condition
Posts: 8617 Joined: 30-Aug-2008 Last visit: 30-May-2026 Location: square root of minus one
|
Quote:there was a small spill that quickly turned blue (first picture). Higher pH extracts (eg CaOH) don't seem to do this One would have to test the response of the blue stain to changes in pH as well. Consider the possibility that the oxidation product turns/remains colourless at higher pH. I'm not saying that it does, but among the possible structures of psilocin oxidation products are some that would show colour changes according to pH. “There is a way of manipulating matter and energy so as to produce what modern scientists call 'a field of force'. The field acts on the observer and puts him in a privileged position vis-à-vis the universe. From this position he has access to the realities which are ordinarily hidden from us by time and space, matter and energy. This is what we call the Great Work." ― Jacques Bergier, quoting Fulcanelli
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
downwardsfromzero wrote:Quote:there was a small spill that quickly turned blue (first picture). Higher pH extracts (eg CaOH) don't seem to do this One would have to test the response of the blue stain to changes in pH as well. Consider the possibility that the oxidation product turns/remains colourless at higher pH. I'm not saying that it does, but among the possible structures of psilocin oxidation products are some that would show colour changes according to pH. Very good point, hadn't considered that I was assuming the enzymes that start the undesriable psylocybin->psilocin->oxidation did not work at higher pH, but we need to keep other possibilities in mind until we really know. Thank you.
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Infundibulum wrote:Loveall wrote:Infundibulum wrote:Have done the calcium bit, does not work
I need to get back to my notes and provide more details.
Many thanks for any info you can find Infundibulum. I'm a fan of your work here as I read back through the years   A few questions if you have the info: Did something precipitate (currently we are just getting goop)? Any cleanup before attempting the precipitation? What was the Ca source? What about the Ca concentration? pH? Temperature? And again, thank you. Thanks  So the details are: 1. Got 5 grams of P.semilanceatas , which have high amounts of psilocybin and traces of psilocin. 2. extracted them with boiling water, 3 x at room temperature. 3. all aqueous extracts were combined and additional water saturated with calcium hydroxide was added to pH of 12. 4. Dark precipitates formed which were left to settle. Supernatant was decanted, dark residue was dried. 5. More calcium hydroxide was added to the decanted supernatant (did not measure pH) and more dark precipitates which were collected as per step #4. 6. Bioassays: -mushroom residue was mildly active, which was expected. Was not aiming on an exhaustive extraction anyway. -dark residues were made into a lemon tea and ingested. Total lack of activity. -Supernatant was newtralised , then drunk. ACtive, but not as active as one would expect frm the equivalent of 5 grams of semilanceatas (1% psilocybin) My verdict is that the calcium precipitates is something else maybe some sort of saccharide, proteins or combination of each. On its own, the dark precipitate does not taste of anything. Any actives remained in the supernatant and my guess is that their ativity was diminished due to prolonged exposure to high pH. There is a study somewhere (maybe even I put it in this thread in the past?) that has to do with isoelectric focussing of psilocybin claiming something along the lines that psilocybin is not as stable at pH>12. I would be more than happy to see those results repeated by others! I'm also finding that the calcium psylocibinate salt does not precipitate. Tried to force the precipitation by adding CaCl2, but Ca(OH)2 starts precipitating, capping the Ca++ concentration at high pH. The Car(OH)2 / CaCl2 / Water system seems complicated (see image below from here), maybe there is a window for psilochbin salt precipitation (bellow freezing, etc). Bottom line: It seems that Ca(OH)2 solubility is not good enough to get this to work to first order. However, it does seem to be a good clarifying agent (and is used as such in for example the sugar industry). Sugar or glycerol seem to enhance Ca(OH)2 solubility and that may be worth looking into. I can't say that I have noticed any loss of activity of what stays in solution due to pH > 12. To minimize degradation I try to work in dim lighting. Maybe pH>12 does degrade psilocybin over time, but I have not noticed this. There may be another path. Na(OH) and NaCl are soluble in alcohol, while Na phosphate salts are not. This means we can try to precipitate sodium psilocybinate in the alcohol with the aid of lye and table salt. May also be worth a try in water too (alkali Manske) since even if sodium phosphates are soluble in water, the NaCl may still be able to crash them out. Loveall attached the following image(s): IMG_20180407_105103.jpg (15kb) downloaded 311 time(s).
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Update on this work. On the most recent test, 260g of fresh mushrooms where put directly in boiling water with excess Ca(OH)2. After reducing/settling/decanting ~150ml of a clear tan liquid is obtained (first image, Ca(OH)2 is indeed a good clarifying agent!). pH is 12 (expected since it should be saturated with Ca(OH)2). Based on previous work, the actives are expected to be in this liquid in the form of psylocybin. Several experiments have been happening (20ml at a time). Want to give an update on one of the splits. When NaOH is added some cloudiness appears (expected from Ca(OH)2 precipitation, see second figure). 20% NaCl was added but nothing happened. However, as more NaOH is added the pH drops and at one point a really thick cloudiness appears which settles after some time in the fridge. pH is still relatively low (~11) at this point. This could simply be salt crashing out (see Figure 3) and proteins. Plan is to keep on adding NaOH to check for more clouds forming. Once we get above pH 12 will consider nenutralizing and bioassying each part or simply running more experiments. The NaOH may destroy the actives, or it may be able to form and push out disodium psylocibynate. I just don't know. NaOH can precipitate NaCl it seems (Figure 3). It may be able to strip off the protons from the phosphate group where Ca(OH)2 can't?. Input welcome. Specifically, anyone know why the pH is dropping when adding NaOH? Are we liberating some protons from phosphates or aminoacids in the extract? Have room for more experiments with the 20ml samples if anyone wants to suggest something. As mentioned before, this can also be tested in alcohol where the disodiun-phosphate to NaOH solutility ration is even lower. Testid this once on a small sample and got some precipitation, but as usual need to test if it is just moar proteins/gunk. Loveall attached the following image(s): IMG_20180408_194109946_LL.jpg (3,751kb) downloaded 296 time(s). IMG_20180409_150034.jpg (44kb) downloaded 297 time(s). IMG_20180409_150006.jpg (61kb) downloaded 294 time(s).
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Adding NaOH and salt directly to the clarified Ca(OH)2 saturated tea does not work. The pH just keeps on dropping (pH went from 12 down to 8.5) until a congealed paste forms. Maybe this is from all the organic matter (proteins, etc) still in the tea (speculation). There are ways to precipitate/separate moar gunk from the clarified tea. See attached for an overview of protein removal techniques. To clean the tea further, I added 3x the volume of acetone. A white cloud formed (first picture). It precipitated to a tan gunk (second picture) and the supernatant decanted. Water with ammonium sulpahte were added to this and two layers formed, the water rich layer (bottom) was recovered. I think water is a lot cleaner than the original tea because: (1) water is clear, (2) adding NaOH now raises the pH (which stars at 7.5 after adding the ammonium sulfate salt) with no signs of congealing, and (3) bubbles quickly pop/fizz out (in the initial tea bubbles would persist a lot moar). I'm hoping the psilochbin (which is not soluble is acetone) has "mostly" stayed in the water rich phase. More updates to follow. Will be trying to force out psilocybinate salt with excess sodium ions in an alkali environment in both water and alcohol (drying the clear water and pulling the dry residue with denatured alcohol should separate any psilochbin from the ammonium sulfate salt and Ca(OH)2 currently in solution). Also plan to add acetone when the pH is at psilocybin's isoelectric point (pH~4) to see if it crashes out. May also be worth revisiting Ca(OH)2 precipitation on the cleaner clear extract. Input or other ideas welcome. Thank you.
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Update: attemps to precipitate sodium psilocybinate salts were unsuccessful. Typically a different salt seems to precipitate first (e.g. Na2SO4 which is pretty water soluble). Fumaric acid seems to be showing some promise so I'll be looking into on that in the near future.
|
|
|
DMT-Nexus member
Posts: 459 Joined: 30-Nov-2012 Last visit: 29-Sep-2025
|
Keep going mate I'm following anyway
|
|
|
analytical chemist

Posts: 7463 Joined: 21-May-2008 Last visit: 09-Aug-2025 Location: the lab
|
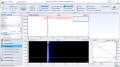
I’m throwing my hat in the ring. will probably do A/B/A, and characterize. this will take a month or so "Nothing is true, everything is permitted." ~ hassan i sabbah
"Experiments are the only means of attaining knowledge at our disposal. The rest is poetry, imagination." -Max Planck
|
|
|

DMT-Nexus member
Posts: 27 Joined: 15-Aug-2013 Last visit: 28-Nov-2023 Location: 192.168.1.1
|
Just spent my last couple of hours dissecting this post and it's all very intriguing! any updates?
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
formulaic wrote:Just spent my last couple of hours dissecting this post and it's all very intriguing! any updates? We were able to get (nor)baeo to crash out from xylene after FASA. However psilocybin remains elusive. There may be a way to get it, but the pH and salt concentration window may be narrow. In the pharmaceutical industry folks adjust these two parameters (along with temperature) to isolate temperamental stuff. The search for a process window seems difficult if one is using bioassays or has to send out stuff for analysis each time. Soooo we are working on a home raman spectrometer setup. If it works one can hopefully track where the psilocybin is real time as salt and pH are adjusted and find a good window where it will move out of water (crash out or go into a solvent). There are a lot of challenges for this (for example, in a crude extract there will be a lot of Spectra overlapping making it too noisy), but we are going to try to proceed this way. One simple option is to try a bufo-like extraction (base/dry/extract with IPA/FASI). Has anyone ever tried something like this?
|
|
|
❤️🔥
Posts: 3648 Joined: 11-Mar-2017 Last visit: 06-Jul-2026 Location: 🌎
|
Expanding the suggestion above with some details: Optional: Wash dry ground mushrooms with 99% IPA vitamin C (this won't pull actives). Dry. Extract mushrooms with calcium hydroxide and a small amount of water (pH should be basic) Decant/Filter Dry and collect gunk Extract with 99% IPA (if it works for bufo...) FASI
|